yl6809永利官网程俊教授课题组在水溶液中电子能级的计算研究取得重要进展,相关成果以“Calculation of Electrochemical Energy Levels in Water Using the Random Phase Approximation and a Double Hybrid Functional”发表在国际物理学期刊Phys. Rev. Lett.(DOI: 10.1103/PhysRevLett.116.086402)。
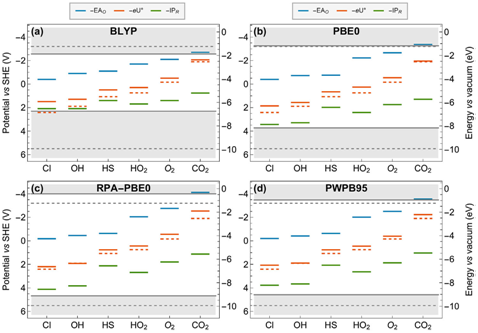
电化学界面极为复杂,为了更好的理解电化学固液界面电荷转移过程,必须将固相和液相(水)的电子结构(包括能带结构、缺陷和溶液离子电子能级)放在同一电子结构理论水平上处理研究。而程俊教授采用近年来课题组发展的基于密度泛函理论的分子动力学模拟(DFTMD)与自由能微扰理论相结合的方法,巧妙的攻克了这一科学难题(Cheng et al. Acc. Chem. Res., 2014, 47, 3522)。但是计算的精度很大程度上局限于密度泛函理论中泛函近似的误差。近期,他们首次采用最新的随机相位近似(random phase approximation)和双杂化泛函(double hybrid)的方法计算了水相中六个氧化还原对的电子能级结构,并把它们和水的电子能级排列在相对于计算标准氢电极的同一能级水平上(如图),计算精度大大提高。这一重要研究进展克服了计算电化学在界面模拟研究上的局限性,能够允许我们精确地将计算结果与实验结果进行分析对比。同时,两种方法都很好的描述了溶质和水的电子能级结构,给予全局的计算研究电化学界面带来了希望。
该研究工作由程俊教授与苏黎世理工大学Joost Vande Vondele教授共同完成。同时,剑桥大学Michiel Sprik教授和复旦大学徐昕教授在对该项目给予了很大帮助。该工作得到国家自然科学基金(Grant No. 21373166)、欧盟第七框架计划(No. 277910.)、瑞士国家基金会(NCCR MARVEL)的资助和支持。
论文链接:http://journals.aps.org/prl/abstract/10.1103/PhysRevLett.116.086402